Datafiles and Scripting¶
Each step of processing and analysis of a genomics pipeline spawns many new files, of many types. Some filetypes, like GFF are only found in a single step of the pipeline, and so are relatively easy to keep track of. However, most are more like FASTQ files, where any given file could be from many different steps of the pipeline. These are the ones that cause the most trouble, and need the most careful management.
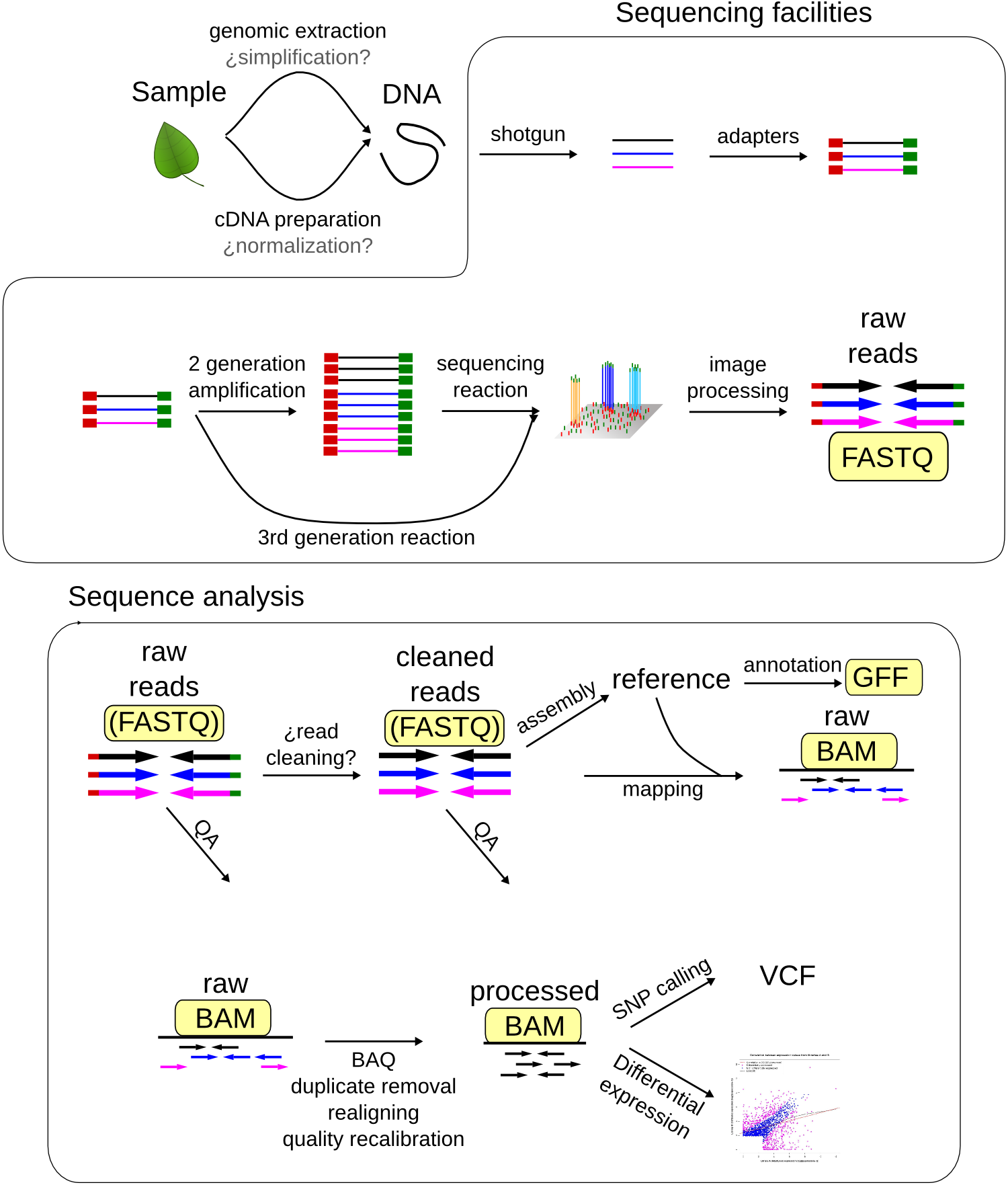
FileTypes
First, lets see what data files we have available:
ls
ls stands for list, and if you call it all by itself, it just
returns a list of whatever is inside the folder you’re currently looking
at. You should give you a fairly big list of files in alphabetical
order. However, they’re hard to understand like this, so lets ask ls
to make the list a little easier to read:
ls -lah
Here we’ve added modifiers to ls. Computer people usually call these
modifiers ‘flags’ or ‘arguments’, and here we’ve added 3 flags: -l
directs ls to give us the results in ‘long format’ so we get more
information -a tells ls to show us ‘all’ of the things in the
folder, even if they’re usually hidden -h makes the output ‘human
readable’, so you see file sizes in kb or gb instead of bytes
SAM and BAM¶
SAM files are tab-delimited files that describe how reads align to a
sequence. They generally start with header lines (which always start
with @) before the actual alignments.
BAM files hold all the same information, but in binary format, which makes them much faster for computers to use, but impossible for us to read. Lets check:
head 12724.bam
head 12724.sam
The .bam file just looks like nonsense, but the .sam file looks
sort of like we expected, except its all headers. So lets look at more
of the SAM file:
head -20 12724.sam
...hmm
head -100 12724.sam
...that’s a lot of headers. Rather than try to guess how far the header goes, lets just look at the other end of the file:
tail -20 12724.sam
tail works just like head, except it counts up from the end of
the file instead of down from the top. So now we can see an example of
the alignment part of the file.The alignments all have at least 11
standard columns (although the values might be zero), but can have lots
of extra ones as well. These are the 11 required columns:
| Col | Field | Type | Brief description |
|---|---|---|---|
| 1 | QNAME | String | Query template NAME |
| 2 | FLAG | Int | bitwise FLAG |
| 3 | RNAME | String | Reference sequence NAME |
| 4 | POS | Int | 1-based leftmost mapping POSition |
| 5 | MAPQ | Int | MAPping Quality |
| 6 | CIGAR | String | CIGAR string |
| 7 | RNEXT | String | Ref. name of the mate/next read |
| 8 | PNEXT | Int | Position of the mate/next read |
| 9 | TLEN | Int | observed Template LENgth |
| 10 | SEQ | String | segment SEQuence |
| 11 | QUAL | String | ASCII of Phred-scaled base QUALity+33 |
Because SAM files are tab-delimited, they are easy for both people and
computers to read, (just not as quickly as BAM files). For instance, we
can use the program cut to get the flags from a SAM file:
cut -f 2 12724.sam
-f which ‘field’ do you want? That was way too much stuff to look
at. So lets make our first script! All we’re going to do it take the
output from tail and send it into cut using a program called
‘pipe’:
tail -20 12724.sam | cut -f 2
Now we have just the flags from the last 20 lines. Instead lets get the flags from the last 20 lines and their sequences:
tail -20 12724.sam | cut -f 2,10
Exercise 1: Get all of the integer type data from the last 30 lines
Exercise 2: Get the quality scores from the penultimate 10 lines
FASTA¶
You’re likely already familiar with FASTA files, as this is the most common way to distribute sequence information. Let’s look at one:
head Raphanus.fa
head is another program, and it shows you just the top few lines of
a file. By default, it shows ten, (so five sequences) but we can also
change that behavior with flags:
head -4 Raphanus.fa
Now, you should see the first four lines of the Raphanus.fa file.
Exercise Try looking at EV813540.fa
FASTA files always have at least one comment line, which almost always begins with “>”, but can start with ”;”. A given sequence in the file is allowed to have multiple comment lines, but they usually don’t. Extra comment lines for sequences can break some downstream processes.
After the comment line is the sequence. Usually this is all on one line, but you can see that this one is formatted so that each sequence line is only 80 characters wide. This makes it easy to read, but makes it slightly more difficult to search within the file. For searching, its nice to have files where all of each sequence is on a single line. For instance, lets see whether there are any EcoRI sites are in the Raphanus.fa file:
grep "GAATTC" Raphanus.fa
grep is a program that searches for any string, and by default returns the entire line that your string is found in. For a file this big, this isn’t very helpful. So lets modify how grep reports it’s findings:
grep -B 1 "GAATTC" Raphanus.fa
-B number grep will return the line with your string plus ‘number’
lines of ‘before context’, so here we’ll get one previous line...the
comment that tells us the sequence name
Now we know which of the sequences have the restriction site we’re looking for, but there’s so many they’ve overfilled the screen. So lets redirect the output from the screen into a file:
grep -B 1 "GAATTC" Raphanus.fa > Raphanus_EcoRI.fa
The greater than sign takes everything that happens on this side of it
> and dumps it into the place designated here. So, all of the output
from that grep command above got saved into a new file called
Raphanus_EcoRI.fa Since we didn’t specify a place to save it, the new
file is just saved in the same folder we’re in, and we can see it by
using ls again:
ls -latr
-r makes the list print to our screen in reverse chronological
order, so the newest files are on the bottom. This makes it easier to
find what we’re looking for.
grep, ls and head all have lots of useful flags, and we can find out what they
are by looking at the manual page:
This opens the manual in the text viewer less, which we’ll talk about more in a few
minutes. For now, the important things to know are that you can scroll line by line
using the arrow keys, or go down one page at a time using the space bar. You can search
for a keyword by typing / and text to search for. Let’s look at the explanation for
a flag we already used:
I actually prefer to look at man pages online, because searching them is easier. Try Googling ‘man grep’
Exercise: How would you changegrep -B 1 "GAATTC" Raphanus.fa > Raphanus_EcoRI.fa
to add line numbers to the output? Hint: [1].
So, now we can make a file that only has sequences with our cut site. Depending on what and why you’re searching, this might be useful for making markers or primers. But maybe we just want to know how many sequences had our cut site:
grep -c "GAATTC" Raphanus.fa
-c grep ‘counted’ 88 instances of EcoRI
Grep happens to have a built in flag for counting matches, but many other programs don’t. So there is a separate program just for counting that we could use by invoking a ‘pipe’:
grep "GAATTC" Raphanus.fa | wc
wc stands for word count, and actually gives us three numbers: number of lines, number
of words and number of characters, in that order. The first two are both 88 because there
are no spaces between the letters of the sequences, so each sequence is interpreted as one
big word.
If we only want one of those numbers, we can use the flags -l, -w, and -c
respectively.
A ‘pipe’ is a little like holding up a real-world pipe, everything you dump in the top
comes out the bottom. Here, the answer from grep "GAATTC" Raphanus.fa goes in and
becomes input for wc. Notice that we only told the computer which file to use for
grep, each pipe after that (there can be an as many as you want) gets its input from
the previous programs output. Also notice that we got rid of all of the grep flags. Why?
Exercise: How would you get just the names of the sequences that match our restriction site? Hint [2]. And save that list to a file?
What if we want to do a ‘fuzzy’ search? Say we want to search for AccI <https://www.neb.com/products/r0161-acci>
which has a recognition sequence of ``GTMKAC` which means GT then either an A
or a C then a G or a T, then AC
Naively, we could search for this cut site by doing a series of greps:
grep "GTAGAC" Raphanus.fa > Raphanus_AccI.fa
grep "GTCGAC" Raphanus.fa > Raphanus_AccI.fa
grep "GTATAC" Raphanus.fa > Raphanus_AccI.fa
grep "GTCTAC" Raphanus.fa > Raphanus_AccI.fa
This has two problems. First, your Raphanus_AccI.fa file will only have results from the fourth grep command, because in each line we’ve save the results as the same file name. That means each time, the previous file is over-written. We can fix that by adding a second print command like this:
grep "GTAGAC" Raphanus.fa > Raphanus_AccI.fa
grep "GTCGAC" Raphanus.fa >> Raphanus_AccI.fa
grep "GTATAC" Raphanus.fa >> Raphanus_AccI.fa
grep "GTCTAC" Raphanus.fa >> Raphanus_AccI.fa
Here, >> means append the results to this file. So now our file will have results from
all four commands. However, we still have the second problem, which is that we’re using our
brains to remember all the combinations bases that match this cut site, but really we
should be making the computer do that. Four lines of code might not seem too arduous, but
consider if you want to look for BglI...it’s recognition sequence is GCCNNNNGGC, which
would take 24 different lines of code. Instead, we’re going to use wildcards. The simplest
wildcards are just brackets that contain the allowed options:
grep "GT[AC][GT]AC" Raphanus.fa > Raphanus_AccI.fa
This gets all four combinations in a single line.
Exercise: grep out just the names for sequences that have a BglI siteGCCNNNNGGCHow many hits are there? Hint [3].
This particular file has all of the sequences in CAPITAL LETTERS, but as we have seen, UNIX is case sensitive. So we get different answers depending on how we phrase our grep:
grep -c "GT[AC][GT]AC" Raphanus.fa
grep -c "gt[ac][gt]ac" Raphanus.fa
Again, naively, we might try:
grep -c "[Gg][Tt][AaCc][GgTt][Aa][Cc]" Raphanus.fa
But this looks like the sort of problem a programmer has already figured out. If we search the grep manual file for ‘case’ we find that we can just tell grep to ignore case:
grep -ci "gt[ac][gt]ac" Raphanus.fa
Note that we can usually bunch up our flags behind a single - so that these two are
exactly the same:
grep -ci "gt[ac][gt]ac" Raphanus.fa
grep -c -i "gt[ac][gt]ac" Raphanus.fa
Lets say that we really will frequently want to look for AccI on all the files in the FASTAS folder. First, lets see whats in there.
cd FASTAS/
ls
ls | wc
less AT1G01060.1
Do we want to type grep -i -B 1 "gt[ac][gt]ac" 25 times? No. Instead we’re going to use
a loop.
A loop is a short program that does the same thing over and over. You just tell it what action you want it to do, and a list of items it should do that action to. It has several important parts:
| for | starts the loop |
| in | sets up the list of items |
| do | sets up the action |
| done | finishes the loop |
Conceptually, we want to tell the computer:
Use the files in FASTAS/, and do grep -i -B 1 "gt[ac][gt]ac" on each one.
for ATfiles in `ls`; do grep -i -B 1 "gt[ac][gt]ac" ${ATfiles} ; done
Notice that there are ‘backticks’ around the ls, backticks are like parentheses is math,
they tell the computer to do that action first.
Exercise: Why does thelshave to get done first??
This does almost what we want, but we’re getting all the sequences, lets just get the name lines:
for ATfiles in `ls`; do grep -i -B 1 "gt[ac][gt]ac" ${ATfiles} | grep ">" ; done
This is even better, and if we wanted all the description information, this would be perfect
but maybe we just want the filenames. Because these files are named for the gene location,
we can get most of the way there by just using cut again.
for ATfiles in `ls`; do grep -i -B 1 "gt[ac][gt]ac" ${ATfiles} | grep ">" | cut -f 1 -d " " ; done
Exercise: Can you figure out how to get rid of the leading > from this list?
Hint [4].
Okay, so this is great, but it’s so complicated. If I want to run this next week, or even tomorrow, I’m never going to remember how we did it. So we’re going to save all this work as a script. Copy that line, then type nano.
Nano is a text editor. Like Word, but in the shell. Paste the line in, then type cntl + o to ‘write out’ and give this file a name, like REscript.sh <enter> It should instantly change colors, that’s ‘syntax highlighting’, the computer has highlighted words it knows to make it easier for you to read the script.
Now we can close nano with cntl + x and we can re-run this script over and over.
sh REscript.sh
We’re doing reproducible science!
Now lets make it better. Reopen the file in nano:
nano REscript.sh
And replace all of the ‘;’ with <return>s, and put a tab before the ‘do’. While we’re at it, lets add a <return> after each pipe as well. What we want is for our script to be readable to us three weeks from now, so lets also add comments. Those are any text that starts with ‘#’. The computer will ignore everything to the right of the ‘#’, and you should fill your scripts with them, you can never have too many comments. You should end up with something like this (Ignore the | at the beginning of each line):
|for ATfiles in `ls`
|do grep -i -B 1 "gt[ac][gt]ac" ${ATfiles} | #search for AccI in a list, get the comment line as well
|grep ">" | #Get only the comment lines
|cut -f 1 -d " " | #Remove the description from the comment lines
|cut -f 2 -d ">" #Remove the leading ">" from the comment lines
|done
Exercise: What would make this script better?
Be able to change search query
Be able to change file list
Have the computer prompt you for input
We made lots of cool variations on this program:
| [1] | Use a -n |
| [2] | You’ll need two greps |
| [3] | either -c or wc should give you the answer to life, the universe and everything |
| [4] | cut will let you use anything as a deliminator |
LICENSE: This documentation and all textual/graphic site content is licensed under the Creative Commons - 0 License (CC0) -- fork @ github. Presentations (PPT/PDF) and PDFs are the property of their respective owners and are under the terms indicated within the presentation.
comments powered by Disqus