MG-RAST and its API¶
Just like the NCBI databases, there are many ways you can interact with MG-RAST, and the web interface is possibly the worst way.
Another way you could work with MG-RAST is to download the entire database and then write parsers to get what you want out of it. I’ve also found this incredibly painful but if you want to do that, you can find its database here.
The best way to access MG-RAST data in my experience is to learn to use their API. MG-RAST has done a decent job publishing API documentation – it just takes a bit of practice to understand its structure.
Example Usage¶
You read a paper, and the authors reference MG-RAST metagenomes. You want to download these so you can reproduce some of the analysis and ask some of your own questions.
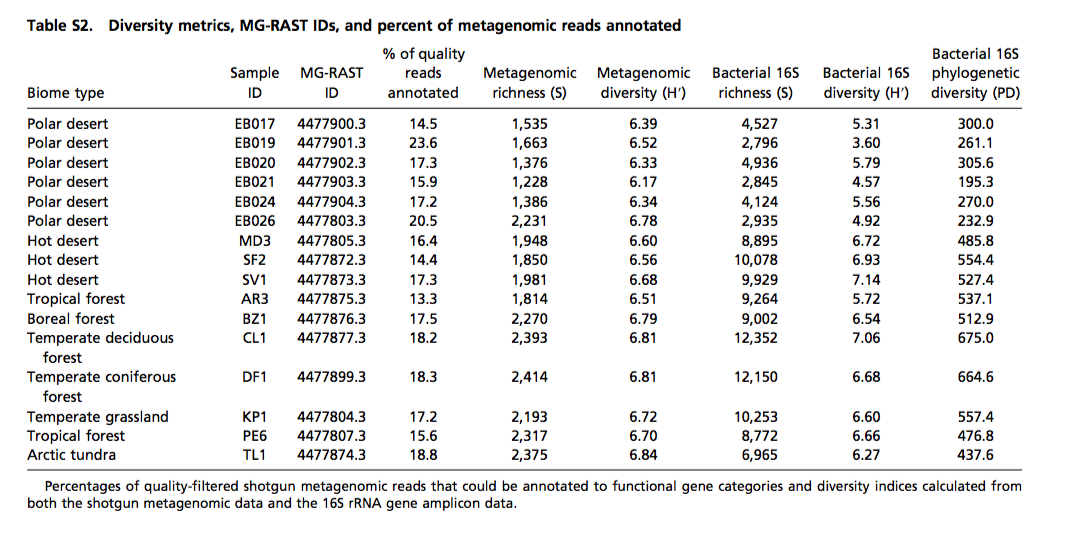
For example, here is some data from a recent PNAS paper, “Cross-biome metagenomic analyses of soil microbial communities and their functional attributes”
If we wanted to download this data with the API, I’d look at the documentation download, here. You’ll see a couple examples that lists how you would download different stages:
http://api.metagenomics.anl.gov/1/download/mgm4447943.3?file=350.1
Or...:
http://api.metagenomics.anl.gov/1/download/mgm4447943.3?stage=650
These two commands above download a specific file or show files from a specific stage for the MG-RAST metagenome ID 4447943.3. You’ll notice how they look similar to the NCBI API calls, with a specific structure. You’re also requesting specific data with the query terms given after the ID with this & structure. Try putting these urls into your web browser and you can see the results.
Remember that you can also access the same commands on the shell with the curl command, but you need to know what kind of output you expect.
This command outputs a file so you need to save the file to an output:
curl “http://api.metagenomics.anl.gov/1/download/mgm4447943.3?file=350.1” > 350.1.fastq.gz
This command returns text (in JSON structure):
As a beginner, I often didn’t know what to expect and would just try things out – which I recommend as a good way to learn.
Even more useful, I think is the following command:
I like to put this in a web browser because it pretty prints the JSON text output. This command above gives all the data that can be obtained from the download call for this metagenome.
A challenge for MG-RAST is that the types of files and the stages aren’t that well-documented. You can get a good guess of what the files and their content from the download page on the web interface, e.g., here. I can tell you from experience that the most important files for me are as follows:
- File 050.2 - This is the unfiltered metagenome that was originally uploaded to MG-RAST
- File 350.2 & 350.3 - These are the protein coding genes (amino acids and nucleotides)
- File 440.1 - These are predicted rRNA sequences (I do not recommend using MG-RAST for sensitive rRNA annotation. It does not use the internal structure of the gene, which other programs appropriately use for classification)
- File 550.1 - This file shows clustered sequences which are 90% identical, to reduce the number of sequences that need to be annotated. Many folks don’t even know that this happens within MG-RAST.
- File 650.1 & 650.2 - These files are essentially the blat tabular output from comparing your sequence to the database.
A few words on the MG-RAST database. This often confuses people about MG-RAST. The central part of the MG-RAST database is a set of known protein sequences. These known sequences are identified by a unique ID (a mix of numbers and letters). Each known sequence is then related to a known annotation in several databases (e.g., RefSeq, KEGG, SEED, etc.). In other words, the search of your sequences to the database involves a sequence comparison to a sequences in the M5nr sequence database and these sequences are then linked to “a hub” of annotations in several databases. If MG-RAST wants to add another database to its capabilities, it would identify the IDs of sequences related to the sequences in the database. If it existed, the new database annotation would be added to the hub. Otherwise, a new ID would be created and also a new annotation hub. As a consequence of all this, the main thing I work with in MG-RAST is these unique IDs.
Exercise - Download¶
Try downloading a few metagenomes from the PNAS paper and associated files. Can you think of how to automate doing this?
MG-RAST annotates sequences and can estimate the abundance of taxonomy and function. Using structured databases like SEED, you can thus find broad functional summaries, e.g., the amount of carbon metabolism in various metagenomes.
In general, I’m paranoid and like to do any sort of abundance counting on my own. Let me give you an example, if one of my sequences hits two sequences in the MG-RAST database with identical scores, what should one do in the abundance accounting?
Working with Annotations¶
Honestly, I’m never sure what MG-RAST is doing, so I like to be in charge of those decisions. Most typically, I am working with 3 types of datasets in any sort of experimental analysis:
- an annotation file linking my sequence to a database (hopefully one with some structure like SEED),
- an abundance file (giving estimates of each of my sequences in my database), and
- some sort of metadata describing my experiment and samples.
MG-RAST can provide you with all three of these, but I typically use it only for #1 (and thus this tutorial also focuses on this). This does require a good deal of know-how in scripting land.
To download these annotation files for specific databases (rather than the unique MG-RAST ID), I use the API annotation command. Using the API, I’ll select the database I’d like to use and the type of data within that database I would like returned (e.g., function, taxonomy, or unique ID – aka md5sum).
There are a couple examples on the documentation that are worth trying:
http://api.metagenomics.anl.gov/1/annotation/sequence/mgm4447943.3?evalue=10&type=organism&source=SwissProt
The above returns a sequence FASTA file with the annotation included in the header of each sequence.:
http://api.metagenomics.anl.gov/1/annotation/similarity/mgm4447943.3?identity=80&type=function&source=KO
I use this more often. The above returns the BLAT results in a tabular format, including the annotations in the last column. Note that with the curl command I can save this to a file and then parse it on my own.
Some comments on the parameters within type within these API calls:
- Organism and function are self-explanatory.
- Ontology is the “structure” of the database, e.g., Subsystems groups SEED sequences into broader functional groups which have their own unique IDS like SS0001.
- Feature - This is the most basic ID within the database of choice, e.g., in RefSeq, this would be its accession ID.
- MD5 - this is the unique ID within MG-RAST.
Note
The other good parameter to be aware of is version. This is important to keep all your analysis consistent. And also guarantees that you are working with the most recent database. Also, when you have to go back and repeat the analysis, you’ll know what version you used. The problem is that MG-RAST has almost no documentation on versions right now. You should write them and complain.
If you do want to download aspects of the database for your analysis, you’ll want to explore the documentation for m5nr API calls. With these calls, you can download the various databases you interact with and more importantly, the ontology structure of databases.
For example, you can see the information for any md5 ID in RefSeq:
http://api.metagenomics.anl.gov//m5nr/md5/000821a2e2f63df1a3873e4b280002a8?source=RefSeq&version=10
Or in all MG-RAST databases:
http://api.metagenomics.anl.gov//m5nr/md5/000821a2e2f63df1a3873e4b280002a8?version=10
If you want to download taxonomy information:
http://api.metagenomics.anl.gov/1/m5nr/taxonomy?version=1
Or functional information in the SEED:
http://api.metagenomics.anl.gov/1/m5nr/ontology?source=Subsystems&min_level=function
Note
One of the things you’ll notice when you run these commands in the command line with curl is that the output is pretty ugly. You’ll want to parse these outputs in a programming language you know and look for a JSON parser. I’m most familiar with Python’s library json, which can import JSON text into Python libraries easily.
I generally use these downloads to link to my annotations. For example, I’d get the SSID that a sequence might be associated with in a BLAT table download and then link it to the database ontology with a m5nr download call.
A note on JSON¶
You might be wondering how to work with these JSON outputs in your own scripting. For example, for this call:
curl http://api.metagenomics.anl.gov//m5nr/md5/000821a2e2f63df1a3873e4b280002a8?version=10
The output of the raw JSON looks like this:

If you look closely, it looks a lot like a Python dictionary structure and that’s how most folks interact with it. Since I program mainly in Python, I use its JSON libraries to work with these outputs in my scripting. I installed the library ijson. In your home directory on your instance, install the library:
wget https://pypi.python.org/packages/source/i/ijson/ijson-1.1.tar.gz
tar -zxvf ijson-1.1.tar.gz
cd ijson-1.1
python setup.py install
You can test that it was installed:
python
>>import ijson
>>
No error message means you’re good to go.
To work with this data structure, I’d look at it first in your pretty JSON-printed webbrowser.
You’ll notice that the data is broken down into a set of nested objects. In this example, the first level contains objects like the version, url, and data. If you go into the data object, you’ll see nested data about source, function, type, ncbi_tax_id, etc.
I access the specific object “data” in Python with the following code:
import urllib
import ijson
url_string = "http://api.metagenomics.anl.gov//m5nr/md5/000821a2e2f63df1a3873e4b280002a8?version=10"
f = urllib.urlopen(url_string)
objects = ijson.items(f, '')
for item in objects:
for x in item["data"]:
print x["function"], x["ncbi_tax_id"], x["organism"], x["source"], x["type"], x["md5"]
Now, if I had a much larger object, say the one below, I’d save it to a file first:
curl http://api.metagenomics.anl.gov/1/m5nr/taxonomy?version=1 > taxonomy_download.json
Then, I would parse through the file:
import urllib
import ijson
import sys
f = open(sys.argv[1])
objects = ijson.items(f, '')
for item in objects:
for x in item["data"]:
if x.has_key("domain"):
print x["domain"], x["ncbi_tax_id"]
#note that not all tax_id's have an associated domain
Exercise - linking MG-RAST to taxonomy¶
One of the most aggravating searches in MG-RAST is linking a md5sum to its taxonomy. But...once you do it, you can give yourself a huge pat on the back for understanding how to interact with this API.
Can you figure out how to do it? For a given md5sum, identify its taxonomic lineage. What if you had to automate this for several md5sums?
- Download the BLAT tabular output for mgm4447943.3 (Hint: the file type is 650.2)
- Identify the best hits for the first 50 reads. (Hint: remember your BLAST tutorial?)
- Find the taxonomy id associated with the first 50 reads using the API call. (Hint: you’re going to want to write your own script for interacting with the following string “http://api.metagenomics.anl.gov//m5nr/md5/” + m5nr + ”?source=GenBank”)
- Find the taxonomy lineage associated with that taxon ID (Hint: See this script.
You can also get taxonomy from NCBI returned in XML format:
http://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=taxonomy&id=376637
Another tool I’ve used is Biopython, which has parsers for XML and Genbank files. Its something I think is worth knowing exists and occasionally I use it, especially for its parsers. Here’s a script that I use it for to get taxonomy for a NCBI Accession Number, here and its also in the repo I’ve been working with during the workshop.
LICENSE: This documentation and all textual/graphic site content is licensed under the Creative Commons - 0 License (CC0) -- fork @ github. Presentations (PPT/PDF) and PDFs are the property of their respective owners and are under the terms indicated within the presentation.
comments powered by Disqus