Variant calling¶
The goal of this tutorial is to show you the basics of variant calling using Samtools.
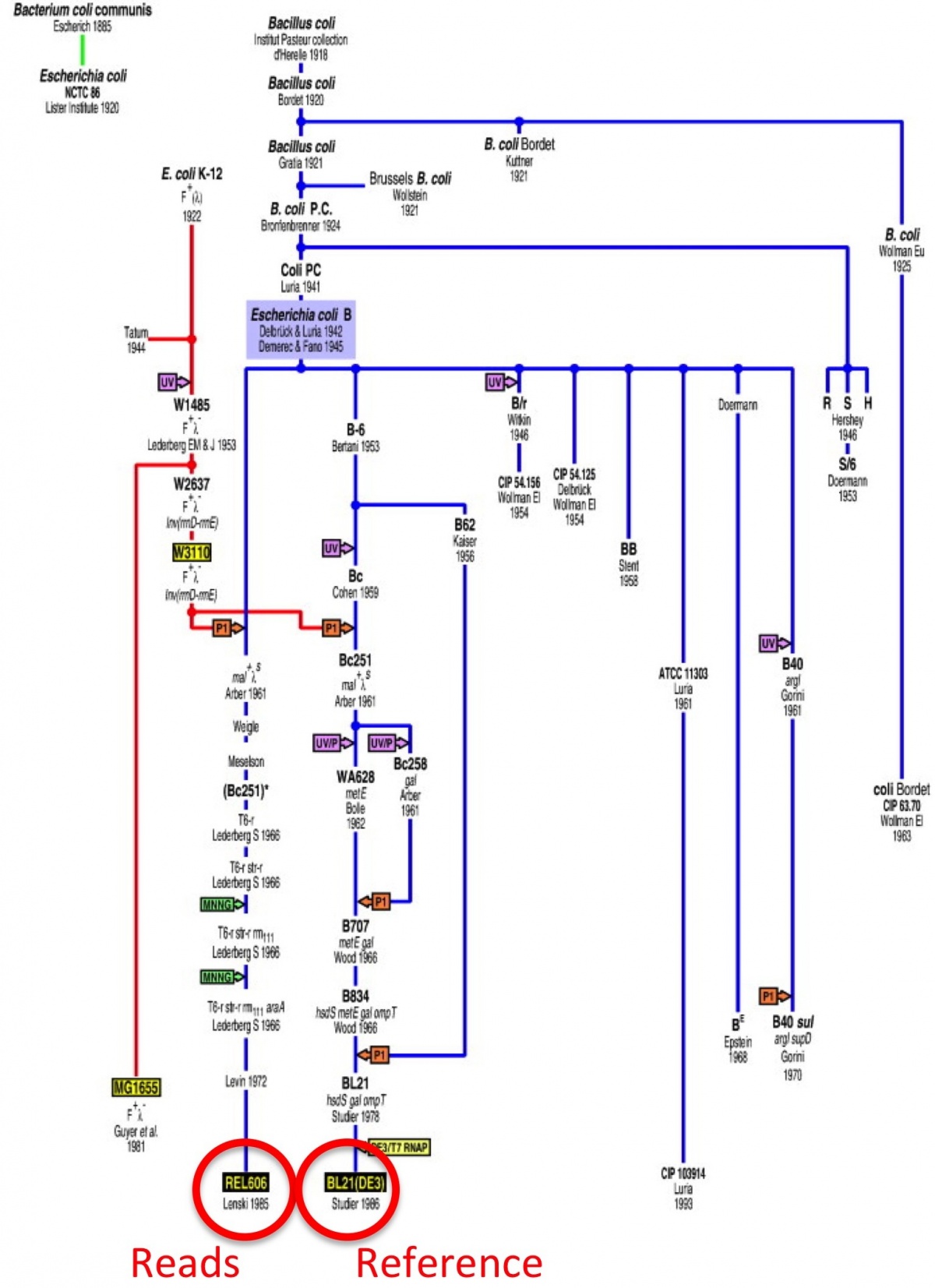
We’re going to be looking at variation in laboratory grown strains of Escherichia coli. We have reads from B strain REL606 and we’ll be mapping it to a reference genome from BL21(DE3). This is a different lab strain, and there’s an interesting paper where they trace the origin and transfer of all the different E. coli strains between scientisits through the decades.
Citation: Tracing Ancestors and Relatives of Escherichia coli B, and the Derivation of B Strains REL606 and BL21(DE3) Journal of Molecular Biology, Volume 394, Issue 4, 11 December 2009, Pages 634–643
Booting an Amazon AMI¶
Start up an Amazon computer (large or xlarge) with an storage of 100Gb.
Install software¶
Log into your instance. Install ruby and git, then install linuxbrew.
sudo apt-get update
sudo apt-get install build-essential
sudo apt-get install ruby git
ruby -e "$(curl -fsSL https://raw.githubusercontent.com/Linuxbrew/install/master/install)"
export PATH="/home/ubuntu/.linuxbrew/bin:$PATH"
export MANPATH="/home/ubuntu/.linuxbrew/share/man:$MANPATH"
export INFOPATH="/home/ubuntu/.linuxbrew/share/info:$INFOPATH"
brew tap homebrew/science
Now we can install anything available from linuxbrew science
brew install samtools
brew install zlib
brew install bcftools
brew install bwa
See what is installed:
brew list
Download data¶
Links to learn more about the data:
Download the reference genome and the resequencing reads
curl "http://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&id=NC_012971&rettype=fasta&retmode=text" > Ecoli_BL21.fasta
curl -O ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR098/SRR098038/SRR098038.fastq.gz
Note, this last URL is the “Fastq files (FTP)” link from the EBI page. Its compressed, lets decompress
gunzip SRR098038.fastq.gz
Just in case EBI is down , you can also get reads this way
curl -O ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/fastq/SRA026/SRA026813/SRX040675/SRR098038.fastq.bz2
Rename the reference¶
The reference is named something really long and complicated. Check it out
head Ecoli_BL21.fasta
Lets shorten that for fewer headaches. Use nano to make the header look like this:
>NC_012971.2
Read mapping¶
Create the BWA index
bwa index Ecoli_BL21.fasta
Now, do the mapping of the raw reads to the reference genome
bwa aln Ecoli_BL21.fasta SRR098038.fastq > SRR098038.sai
Make a SAM file (this would be done with ‘sampe’ if these were paired-end reads)
bwa samse Ecoli_BL21.fasta SRR098038.sai SRR098038.fastq > SRR098038.sam
A sam file contains all of the information about where each read hits on the reference. Links for more info:
Next, index the reference genome with samtools
samtools faidx Ecoli_BL21.fasta
Convert the SAM into a BAM file
samtools view -bS SRR098038.sam > SRR098038.bam
Sort the BAM file
samtools sort SRR098038.bam > SRR098038.sorted.bam
And index the sorted BAM file
samtools index SRR098038.sorted.bam
Visualizing alignments¶
At this point you can visualize with samtools tview. Other visualization software: * Tablet. * IGV
‘samtools tview’ is a text interface that you use from the command line; run it like so
samtools tview SRR098038.sorted.bam Ecoli_BL21.fasta
The ‘.’s are places where the reads align perfectly in the forward direction, and the ‘,’s are places where the reads align perfectly in the reverse direction. Mismatches are indicated as A, T, C, G, etc.
You can scroll around using left and right arrows; to go to a specific coordinate, use ‘g’ and then type in the contig name and the position. For example, type ‘g’ and then ‘NC_012971.2:553093<ENTER>’ to go to position 553093 in the BAM file. (This name is taken from the fasta reference file, you could change to something more reasonable).
Use ‘q’ to quit.
Statistics of alignments¶
This command
samtools view -c -f 4 SRR098038.bam
will count how many reads DID NOT align to the reference (214518).
This command
samtools view -c -F 4 SRR098038.bam
will count how many reads DID align to the reference (6832113).
And this command
wc -l SRR098038.fastq
will tell you how many lines there are in the FASTQ file (28186524). Reminder: there are four lines for each sequence.
There is another package, Picard Tools, that can give you more in depth information. Lets install with linuxbrew
brew install picard-tools
And use the particular tool CollectAlignmentSummaryMetrics
picard CollectAlignmentSummaryMetrics R=Ecoli_BL21.fasta I=SRR098038.sorted.bam O=statistics.txt
You can see the output with cat
cat statistics.txt
Calling SNPs¶
You can use samtools and bcftools to call SNPs. They have great documentation of a standard workflow for calling SNPs, you should read more about it. We’re going to do a simplified and updated version here.
Start with mpileup and pipe the results to bcftools
samtools mpileup -uf Ecoli_BL21.fasta SRR098038.sorted.bam | bcftools call -vmO v -o SRR098038.vcf --ploidy 1 --threads 2
You can check out the VCF file by using ‘tail’ to look at the bottom
tail SRR098038.vcf
Each variant call line consists of the chromosome name (for E. coli REL606, there’s only one chromosome); the position within the reference; an ID (here always ‘.’); the reference call; the variant call; and a bunch of additional information about the variant.
The information at the end can be very useful but difficult to interpret. One way to quickly look up the label shorthand is to grep
grep '<ID=VDB' SRR098038.vcf
grep '<ID=AC' SRR098038.vcf
samtools tview SRR098038.sorted.bam Ecoli_BL21.fasta
You can use ‘samtools tview’ again and then type (for example) ‘g’ ‘rel606:4616538’ to go visit one of the positions. The format for the address to go to with ‘g’ is ‘chr:position’.
NC_012971.2:4558366
You can read more about the VCF file format here.
Using IGV for Visualization¶
Installing IGV requires registration and some patience. IGV Link.
To open your alignments, you’ll need
three files on your local computer: Ecoli_BL21.fasta, SRR098038.sorted.bam,
and SRR098038.sorted.bam.bai. You can copy them over using scp (secure copy),
for example. You will do this from a terminal on your computer that is NOT connected to amazon.
scp -i ~/Downloads/???.pem ubuntu@???:/home/ubuntu/Ecoli_BL21.fasta ~/Downloads
scp -i ~/Downloads/???.pem ubuntu@???:/home/ubuntu/SRR098038.sorted.bam ~/Downloads
scp -i ~/Downloads/???.pem ubuntu@???:/home/ubuntu/SRR098038.sorted.bam.bai ~/Downloads
To add the gene annotation, get this file as well
curl ftp://ftp.ncbi.nlm.nih.gov/genomes/archive/old_refseq/Bacteria/Escherichia_coli_BL21_DE3__uid161947/NC_012971.gff
Student Exercise¶
You are eager to use some E. coli reads from a collaborator, which you can download here
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR201/004/SRR2014554/SRR2014554_1.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR201/004/SRR2014554/SRR2014554_2.fastq.gz
You need to quality trim them, map them to the E. coli reference, and call SNPs. How far can you get?
LICENSE: This documentation and all textual/graphic site content is licensed under the Creative Commons - 0 License (CC0) -- fork @ github. Presentations (PPT/PDF) and PDFs are the property of their respective owners and are under the terms indicated within the presentation.
comments powered by Disqus