15. Annotating and evaluating a de novo transcriptome assembly¶
At the end of this lesson, you will be familiar with:
- how to annotate a de novo transcriptome assembly
- parse GFF3 output from the annotation output to use for DE analysis
- several methods for evaluating the completeness of a de novo transcriptome assembly
- What a Jupyter notebook is and how to execute a few commands in Python
15.1. Annotation with dammit¶
dammit!
dammit is an annotation pipeline written by Camille Scott. The dammit pipeline runs a relatively standard annotation protocol for transcriptomes: it begins by building gene models with Transdecoder, then uses the following protein databases as evidence for annotation:
Pfam-A, Rfam, OrthoDB, uniref90 (uniref is optional with--full).
If a protein dataset for your organism (or a closely-related species) is available, this can also be supplied to the dammit pipeline with the --user-databases as optional evidence for the annotation.
In addition, BUSCO v3 is run, which will compare the gene content in your transcriptome with a lineage-specific data set. The output is a proportion of your transcriptome that matches with the data set, which can be used as an estimate of the completeness of your transcriptome based on evolutionary expectation (Simho et al. 2015).
There are several lineage-specific datasets available from the authors of BUSCO. We will use the metazoa dataset for this transcriptome.
15.1.1. Installation¶
Annotation necessarily requires a lot of software! dammit attempts to simplify this and make it as reliable as possible, but we still have some dependencies.
Create a python 3 environment for dammit:
conda create -y --name py3.dammit python=3
Then
source activate py3.dammit
dammit can be installed via bioconda. Due to some dependency issues with bioconda packages, first run:
conda config --add pinned_packages 'r-base >=3.4'
Add the appropriate channels, including bioconda:
conda config --add channels defaults
conda config --add channels conda-forge
conda config --add channels bioconda
Then, you can install dammit normally (this will take some time, ~5-10 min):
conda install -y dammit
To make sure your installation was successful, run
dammit help
This will give a list of dammit’s commands and options:
usage: dammit [-h] [--debug] [--version] {migrate,databases,annotate} ...
dammit: error: invalid choice: 'help' (choose from 'migrate', 'databases', 'annotate')
The version (dammit --version) should be:
dammit 1.0rc2
15.1.1.1. Database Preparation¶
dammit has two major subcommands: dammit databases and dammit annotate. The databases command checks that databases are installed and prepared, and if run with the --install flag,
it will perform that installation and preparation. If you just run dammit databases on its own, you should get a notification that some database tasks are not up-to-date. So, we need
to install them!
Note: if you have limited space on your instance, you can also install these databases in a different location (e.g. on an external volume). Run this command before running the database install.
#Run ONLY if you want to install databases in different location.
#To run, remove the `#` from the front of the following command:
# dammit databases --database-dir /path/to/databases
Install databases (this will take a long time, usually >10 min):
dammit databases --install --busco-group metazoa
We used the “metazoa” BUSCO group. We can use any of the BUSCO databases, so long as we install them with the dammit databases subcommand. You can see the whole list by running
dammit databases -h. You should try to match your species as closely as possible for the best results. If we want to install another, for example:
dammit databases --install --busco-group protists
Phew, now we have everything installed!
Now, let’s take a minute and thank Camille for making this process easy for us by maintaining a recipe on bioconda. This saves us a lot of hassle with having to install individual parts required for the pipeline. AND on top of the easy installation, there is this slick pipeline! Historically, transcriptome annotation involved many tedious steps, requiring bioinformaticians to keep track of parsing databases alignment ouptut and summarizing across multiple databases. All of these steps have been standardized in the dammit pipeline, which uses the pydoit automation tool. Now, we can input our assembly fasta file -> query databases -> and get output annotations with gene names for each contig - all in one step. Thank you, Camille!
15.1.2. Annotation¶
Keep things organized! Let’s make a project directory:
cd ~/
mkdir -p ~/annotation
cd ~/annotation
You all ran Trinity last week to generate an assembly. The output from Trinity is a file, Trinity.fasta. Today, we’re going to use a de novo transcriptome assembly from Nematostella vectensis.
curl -OL https://darchive.mblwhoilibrary.org/bitstream/handle/1912/5613/Trinity.fasta
head -3000 Trinity.fasta > trinity.nema.fasta
Now we’ll download a custom Nematostella vectensis protein database. Somebody has already created a proper database for us Putnam et al. 2007 (reference proteome available through uniprot). If your critter is a non-model organism, you will likely need to grab proteins from a closely-related species. This will rely on your knowledge of your system!
curl -LO ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/reference_proteomes/Eukaryota/UP000001593_45351.fasta.gz
gunzip -c UP000001593_45351.fasta.gz > nema.reference.prot.faa
rm UP000001593_45351.fasta.gz
Run the command:
dammit annotate trinity.nema.fasta --busco-group metazoa --user-databases nema.reference.prot.faa --n_threads 4
While dammit runs, it will print out which task it is running to the terminal. dammit is written with a library called pydoit, which is a python workflow library similar to GNU Make. This not only helps organize the underlying workflow, but also means that if we interrupt it, it will properly resume!
After a successful run, you’ll have a new directory called trinity.nema.fasta.dammit. If you
look inside, you’ll see a lot of files:
ls trinity.nema.fasta.dammit/
Expected output:
annotate.doit.db trinity.nema.fasta.dammit.namemap.csv trinity.nema.fasta.transdecoder.pep
dammit.log trinity.nema.fasta.dammit.stats.json trinity.nema.fasta.x.nema.reference.prot.faa.crbl.csv
run_trinity.nema.fasta.metazoa.busco.results trinity.nema.fasta.transdecoder.bed trinity.nema.fasta.x.nema.reference.prot.faa.crbl.gff3
tmp trinity.nema.fasta.transdecoder.cds trinity.nema.fasta.x.nema.reference.prot.faa.crbl.model.csv
trinity.nema.fasta trinity.nema.fasta.transdecoder_dir trinity.nema.fasta.x.nema.reference.prot.faa.crbl.model.plot.pdf
trinity.nema.fasta.dammit.fasta trinity.nema.fasta.transdecoder.gff3
trinity.nema.fasta.dammit.gff3 trinity.nema.fasta.transdecoder.mRNA
The most important files for you are trinity.nema.fasta.dammit.fasta,
trinity.nema.fasta.dammit.gff3, and trinity.nema.fasta.dammit.stats.json.
If the above dammit command is run again, there will be a message:
**Pipeline is already completed!**
15.1.3. Parse dammit output¶
Cammille wrote dammit in Python, which includes a library to parse gff3 dammit output. To send this output to a useful table, we will need to open the Python environment.
To do this, we will use a Jupyter notebook. In addition to executing Python commands, Jupyter notebooks can also run R (as well as many other languages). Similar to R markdown (Rmd) files, Jupyter notebooks can keep track of code and output. The output file format for Jupyter notebooks is .ipynb, which GitHub can render. See this gallery of interesting Jupyter notebooks.
Install Jupyter notebook:
pip install jupyter
Then
jupyter notebook --generate-config
Then generate a config file. (Note: this password protects the notebook.)
cat >> ~/.jupyter/jupyter_notebook_config.py <<EOF
c = get_config()
c.NotebookApp.ip = '*'
c.NotebookApp.open_browser = False
c.NotebookApp.password = u'sha1:d3f13af9db69:31268fb729f127aebb2f77f7b61fa92d6c9e3aa1'
c.NotebookApp.port = 8000
EOF
Now run the jupyter notebook:
jupyter notebook &
You will see a list of files, start a new Python 3 notebook:
This will open a new notebook.
Enter this into the first cell:
import pandas as pd
from dammit.fileio.gff3 import GFF3Parser
Press Shift + Enter to execute the cell.
To add a new cell, with the “plus” icon.
In a new cell enter:
gff_file = "trinity.nema.fasta.dammit/trinity.nema.fasta.dammit.gff3"
annotations = GFF3Parser(filename=gff_file).read()
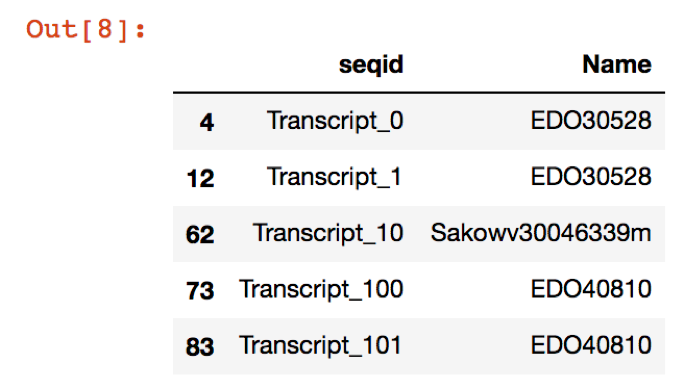
names = annotations.sort_values(by=['seqid', 'score'], ascending=True).query('score < 1e-05').drop_duplicates(subset='seqid')[['seqid', 'Name']]
new_file = names.dropna(axis=0,how='all')
new_file.head()
Which will give an output that looks like this:
Try commands like,
annotations.columns
and
annotations.head()
or
annotations.head(50)
**Questions
- What do these commands help you to see?
- How might you use this information to modify the
namesline in the code above?
To save the file, add a new cell and enter:
new_file.to_csv("nema_gene_name_id.csv")
Now, we can return to the terminal, Control + C to cancel and close the Jupyter notebook.
We can look at the output we just made, which is a table of genes with ‘seqid’ and ‘Name’ in a .csv file: nema_gene_name_id.csv.
less nema_gene_name_id.csv
Notice there are multiple transcripts per gene model prediction. This .csv file can be used in tximport in downstream DE analysis.
15.2. Evaluation¶
We will be using Transrate and Busco!
15.2.1. Transrate¶
Transrate serves two main purposes. It can compare two assemblies to see how similar they are. Or, it can give you a score which represents proportion of input reads that provide positive support for the assembly. Today, we will use transrate to compare two assemblies. To get a transrate score, we would need to use the trimmed reads, which takes a long time. For a further explanation of metrics and how to get a transrate score, see the documentation and the paper by Smith-Unna et al. 2016.
15.2.1.1. Install Transrate¶
cd
curl -SL https://bintray.com/artifact/download/blahah/generic/transrate-1.0.3-linux-x86_64.tar.gz | tar -xz
cd transrate-1.0.3-linux-x86_64
./transrate --install-deps ref
rm -f bin/librt.so.1
echo 'export PATH="$HOME/transrate-1.0.3-linux-x86_64":$PATH' >> ~/.bashrc
source ~/.bashrc
conda activate py3.dammit
- How do the two transcriptomes compare with each other?
cd ~/annotation
transrate --reference=Trinity.fasta --assembly=trinity.nema.fasta --output=subset_v_full
transrate --reference=trinity.nema.fasta --assembly=Trinity.fasta --output=full_v_subset
The results will be in two separate directoreis, with the important metrics in the assemblies.csv files.
cat full_v_subset/assemblies.csv
cat subset_v_full/assemblies.csv
15.2.2. BUSCO¶
- Metazoa database used with 978 genes
- “Complete” lengths are within two standard deviations of the BUSCO group mean length
- Useful links:
- Website: http://busco.ezlab.org/
- Paper: Simao et al. 2015
- User Guide
15.2.2.1. Run the command:¶
We’ve already installed and ran the BUSCO command with the dammit pipeline. Let’s take a look at the results.
Check the output:
cat trinity.nema.fasta.dammit/run_trinity.nema.fasta.metazoa.busco.results/short_summary_trinity.nema.fasta.metazoa.busco.results.txt
- Challenge: How do the BUSCO results of the full transcriptome compare?
Run the BUSCO command by itself:
run_BUSCO.py \
-i trinity.nema.fasta \
-o nema_busco_metazoa -l ~/.dammit/databases/busco2db/metazoa_odb9 \
-m transcriptome --cpu 4
When you’re finished, exit out of the conda environment:
source deactivate