13. Mapping and variant calling on yeast transcriptome¶
Learning objectives:
define and explore the concepts and implications of shotgun sequencing;
explore coverage;
understand the basics of mapping-based variant calling;
learn basics of actually calling variants & visualizing.
13.1. Boot up a Jetstream¶
You should still have your jetstream instance running, you can following the instructions here to log in to JetStream and find your instance. Then ssh into it following the instructions here.
13.2. Install software¶
conda install -y bwa samtools bcftools
13.3. Change to a new working directory and map data¶
After installing the necessary software, we will create the working directory for the mapping as follows:
cd ~/
mkdir mapping
cd mapping
Next, we will create links from the previously downloaded and quality-trimmed yeast dataset:
ln -fs ~/quality/*.qc.fq.gz .
ls
13.4. Variant Calling Workflow¶
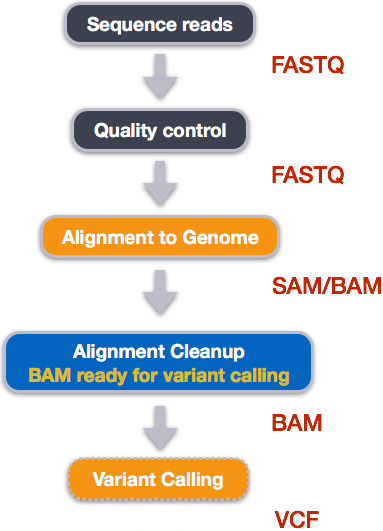
13.5. Map data¶
Goal: perform read alignment (also known as mapping) to determine where our reads originated from.
13.5.1. Download and gunzip the reference:¶
Here we are going to align our transcripts to the reference’s open reading frames to look for single-nucleotide variants. It’s important to think about what reference is appropriate for your experiment. Many biologically important variants exist in non-coding regions, so if we were looking at genomic sequences, it would be important to use a different reference such as the whole genome.
curl -O https://downloads.yeastgenome.org/sequence/S288C_reference/orf_dna/orf_coding.fasta.gz
gunzip orf_coding.fasta.gz
Let’s take a look at our reference:
head orf_coding.fasta
13.5.2. Finding Help with bwa¶
To bring up the manual page for bwa we can type:
bwa
Notice that it brings up a description of the different commands within the bwa software. We will be using index and mem.
We can also have a look at the bwa options page.
13.5.3. Indexing: Prepare reference for mapping:¶
Our first step is to index the reference sequences for use by BWA. Indexing allows the aligner to quickly find potential alignment sites for query sequences in a reference, which saves time during alignment. Indexing the reference only has to be run once. The only reason you would want to create a new index is if you are working with a different reference or you are using a different tool for alignment.
bwa index orf_coding.fasta
13.5.4. Mapping¶
We use an algorithm called bwa mem to perform mapping. To find more information via the help page for the bwa mem function we can type the command below. Without any input, the help page will come up by default:
bwa mem
Then, when we are ready perform mapping with our sample ERR458493 we can type:
bwa mem -t 4 orf_coding.fasta ERR458493.qc.fq.gz > ERR458493.sam
While we are running bwa with the default parameters here, your use case might require a change of parameters. NOTE: Always read the manual page for any tool before using and make sure the options you use are appropriate for your data.
What is the difference between Salmon and bwa mem?
Standard alignment tools (Hisat2, STAR, BWA) try to find the read origin by FULL-ALIGNMENT of reads to a genome or transcriptome.
Ultra-fast alignment-free methods, such as Sailfish, Salmon and Kallisto, have been developed by exploiting the idea that precise alignments are not required to assign reads to their origins
Salmon’s “quasi-mapping” approach uses exact matching of k-mers (sub-strings in reads) to approximate which read a transcipt originated from. The idea behind it being that it may not be important to exactly know where within a transcript a certain read originated from. Instead, it may be enough to simply know which transcript the read represents.
Salmon therefore does not generate a BAM file because it does not worry about finding the best possible alignment. Instead, it yields a (probabilistic) measure of how many reads originated from each transcript. This is enough information for read quantification, and is really fast.
However, BWA
memproduces an alignment, where an entire read is mapped exactly against a reference sequence. This produces more information that is important for things like variant calling.
13.5.5. Observe!¶
13.5.6. SAM/BAM File formats¶
We can peek at our “.sam” file:
head ERR458493.sam
tail ERR458493.sam
The SAM file is a tab-delimited text file that contains information for each individual read and its alignment to the reference. While we do not have time to go in detail of the features of the SAM format, the paper by Heng Li et al. provides a lot more detail on the specification.
The compressed binary version of SAM is called a BAM file. We use this version to reduce size and to allow for indexing, which enables efficient random access of the data contained within the file.
PRACTICE! Using the
bwacommand we ran above as the foundation, construct a for loop to generate “.sam” alignment files for all of our quality controlled samples!Solutionfor filename in *.qc.fq.gz do name=$(basename $filename .qc.fq.gz) echo Working on: $name bwa mem -t 4 orf_coding.fasta $filename > ${name}.sam done
13.6. Visualize mapping¶
Goal: make it possible to vizualize some of our mapping results.
13.6.1. Index the reference:¶
Before we indexed the reference for BWA, now we need to index the reference for samtools. To see the manual for samtools we can type:
samtools
Although both tools use different indexing methods, they both allow the tools to find specific sequences within
the reference quickly. We can see that an indexing function is samtools faidx.
samtools faidx orf_coding.fasta
13.6.2. Convert the SAM file into a BAM file:¶
Next, we will convert our file format to a .bam file with the samtools view command. Let’s see the different parameters for this function:
samtools view
We can see that:
-S: ignored (input format is auto-detected)-b: output BAM
So let’s convert our file format for sample ERR458493:
samtools view -S -b ERR458493.sam > ERR458493.bam
13.6.3. Sort the BAM file by position in genome:¶
You can sort on many different columns within a sam or bam file. After mapping, our
files are sorted by read number. Most programs require mapping files to be sorted by
position in the reference. You can sort a file using the samtools sort command.
samtools sort ERR458493.bam -o ERR458493.sorted.bam
13.6.4. Index the BAM file so that we can randomly access it quickly:¶
samtools index ERR458493.sorted.bam
13.6.5. Call variants with bcftools!¶
Goal: find places where the reads are systematically different from the reference.
A variant call is a conclusion that there is a nucleotide difference vs. some reference at a given
position in an individual genome or transcriptome, often referred to as a Single Nucleotide Polymorphism (SNP).
The call is usually accompanied by an estimate of variant frequency and some measure of confidence. Similar
to other steps in this workflow, there are number of tools available for variant calling. In this workshop
we will be using bcftools, but there are a few things we need to do before actually calling the variants.
First, let’s look at the manual page for bcftools, making note of the mpileup and call commands:
bcftools
Let’s see the different parameters for the bcftools mpileup command by typing:
bcftools mpileup
Note: The parameters we will use for bcftools mpileup are:
-O: Output file type, which can be:‘b’ compressed BCF. We will use this output format!
‘u’ uncompressed BCF
‘z’ compressed VCF
‘v’ uncompressed VCF
-f: faidx indexed reference sequence file
And to find help for the bcftools call function:
bcftools call
Note: The parameters for bcftools call that we will use:
-m: alternative model for multiallelic and rare-variant calling (conflicts with -c)-v: output variant sites only-o: write output to a file
Now, let’s go ahead and run our command on our sample ERR458493!
bcftools mpileup -O b -f orf_coding.fasta ERR458493.sorted.bam | \
bcftools call -m -v -o variants.vcf
Next, we will use a perl script from samtools called vcfutils.pl that will filter out our variants and
we can write the output to a new file.
vcfutils.pl varFilter variants.vcf > variants_filtered.vcf
To look at the entire variants.vcf file you can do cat variants.vcf; all of the lines starting with # are comments. You
can use tail variants.vcf to see the last ~10 lines, which should
be all of the called variants.
The first few columns of the VCF file represent the information we have about a predicted variation:
| column | info |
|---|---|
| CHROM | contig location where the variation occurs |
| POS | position within the contig where the variation occurs |
| ID | a . until we add annotation information |
| REF | reference genotype (forward strand) |
| ALT | sample genotype (forward strand) |
| QUAL | Phred-scaled probablity that the observed variant exists at this site (higher is better) |
| FILTER | a . if no quality filters have been applied, PASS if a filter is passed, or the name of the filters this variant failed |
The Broad Institute’s VCF guide is an excellent place to learn more about VCF file format.
13.6.6. Visualize with tview:¶
Now that we know the locations of our variants, let’s view them with samtools!
Samtools implements a very simple text alignment viewer called tview. This alignment viewer works with short indels and shows MAQ consensus. It uses different colors to display mapping quality or base quality, subjected to users’ choice. Samtools viewer is known to work with an 130 GB alignment swiftly. Due to its text interface, displaying alignments over network is also very fast.
In order to visualize our mapped reads we use tview, giving it the sorted bam file and the reference file:
samtools tview ERR458493.sorted.bam orf_coding.fasta
tview commands of relevance:
left and right arrows scroll
qto quitCTRL-h and CTRL-l do “big” scrolls
Typing
gallows you to go to a specific location, in this format chromosome:location. Here are some locations you can try out:YLR162W:293(impressive pileup, shows two clear variants and three other less clear)YDR034C-A:98(impressive pileup, shows two clear variants)YDR366C:310(impressive pileup, less clear variants)YLR256W:4420(impressive pileup, less clear variants)YBL105C:2179(less depth, shows two clear variants)YDR471W:152(impressive pileup, shows one clear variant)
Get some summary statistics as well:
samtools flagstat ERR458493.sorted.bam